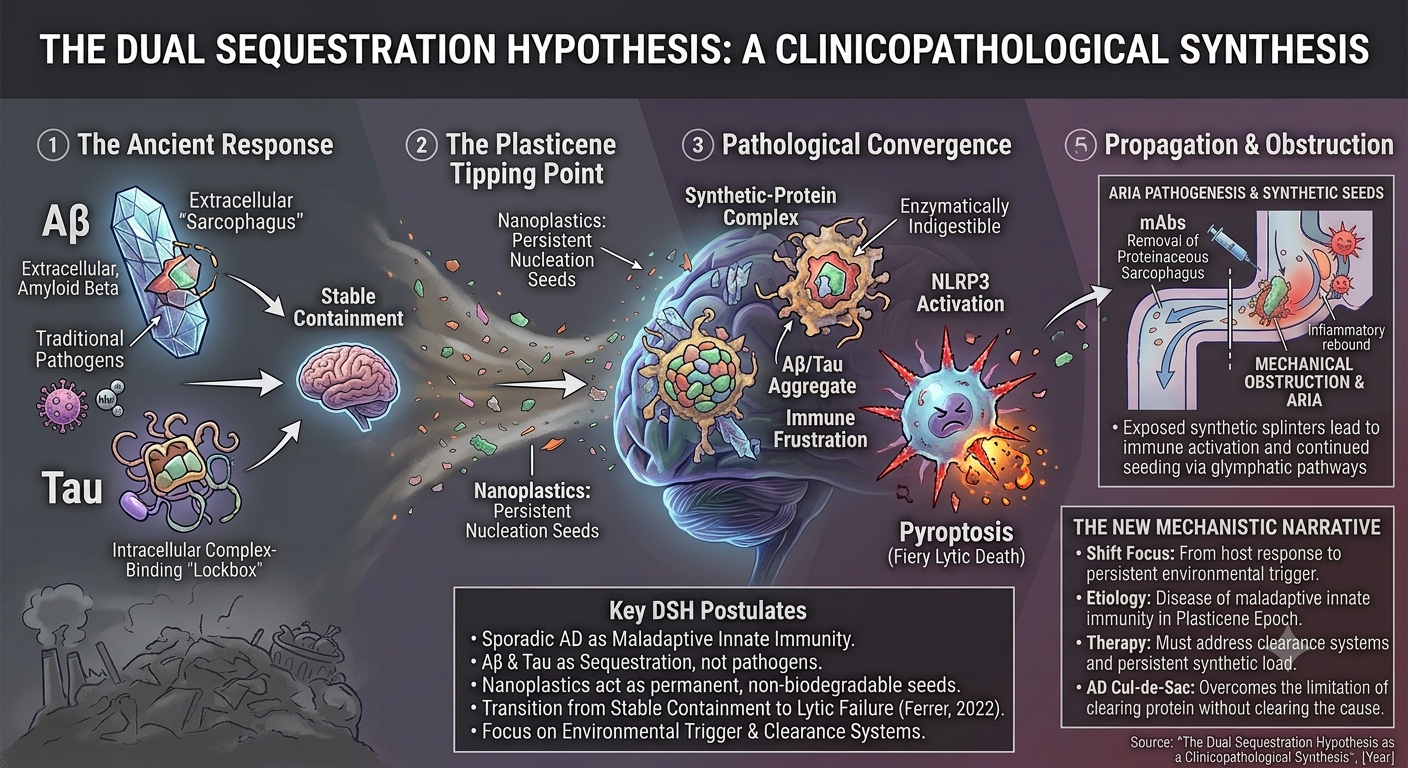
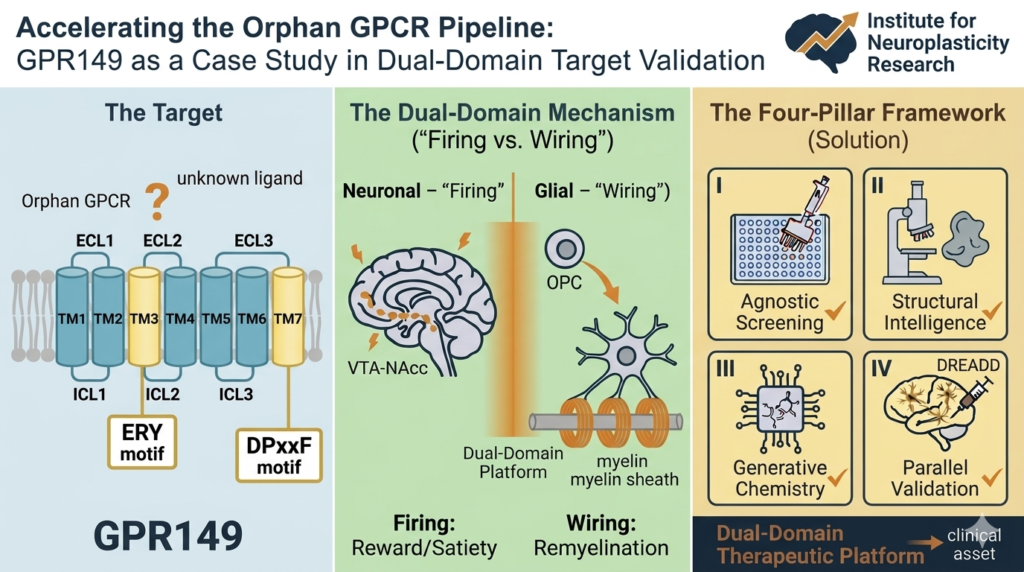
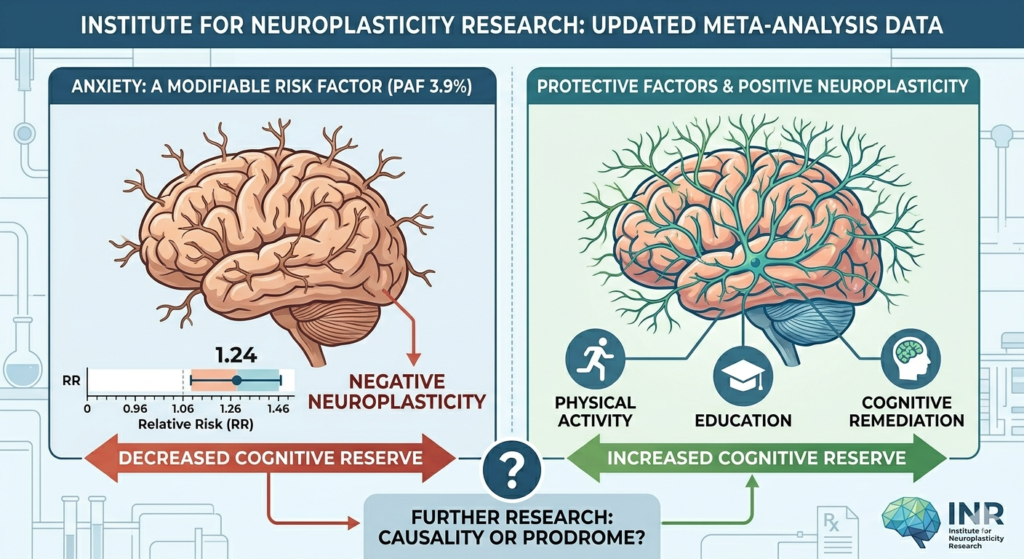

We propose the DSH as a unifying framework to resolve these converging crises. This clinicopathological update posits that sporadic AD, particularly in its modern manifestation, could be understood as a disease of maladaptive innate immunity.
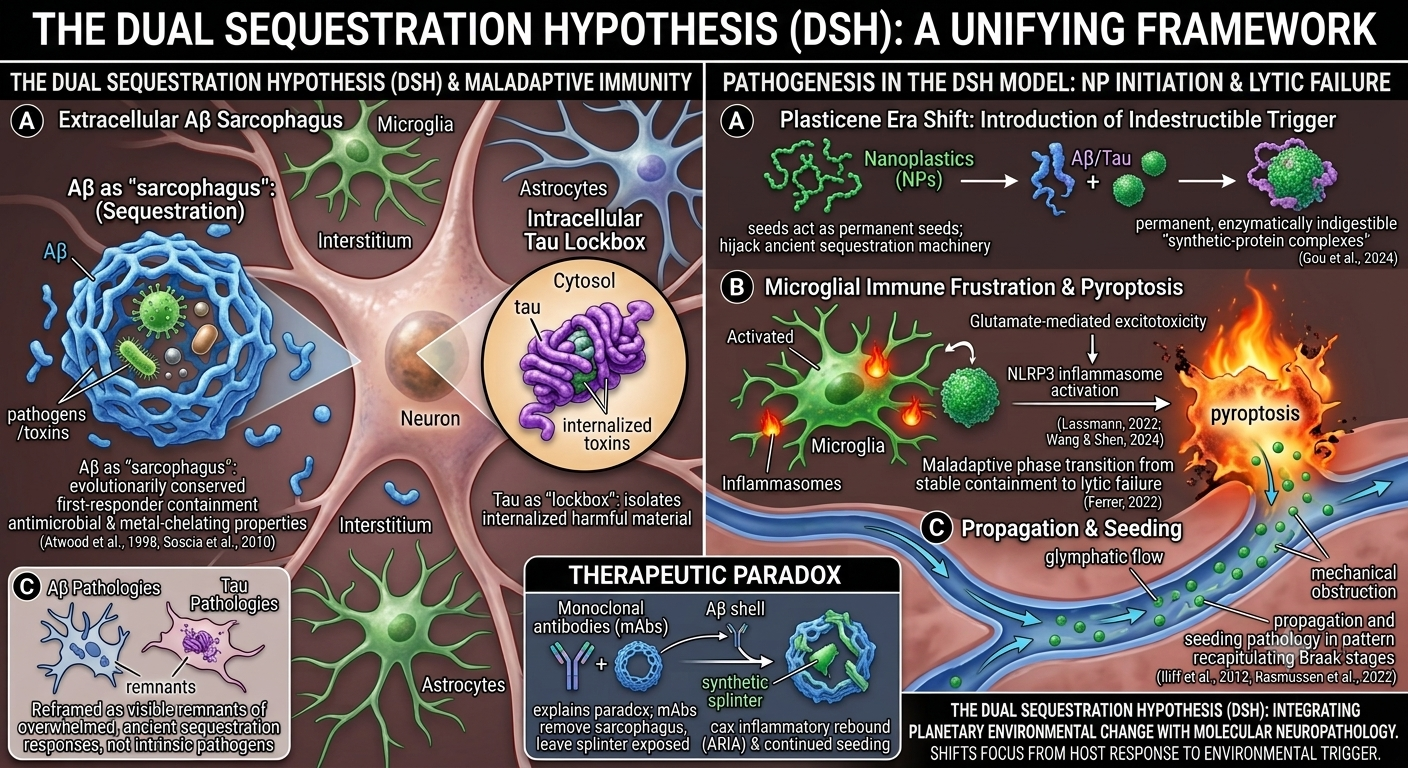
The DSH suggests reframing Aβ and tau pathologies not as intrinsic pathogens but as visible remnants of overwhelmed, evolutionarily conserved sequestration responses. The DSH does not deny that Aβ and tau can exert toxicity in excess or that alternative views regarding their primary pathogenicity have merit. Rather, it reframes their aggregation as an evolutionarily conserved containment response—one that becomes maladaptive when the brain faces an indestructible trigger for which no evolutionary precedent exists.
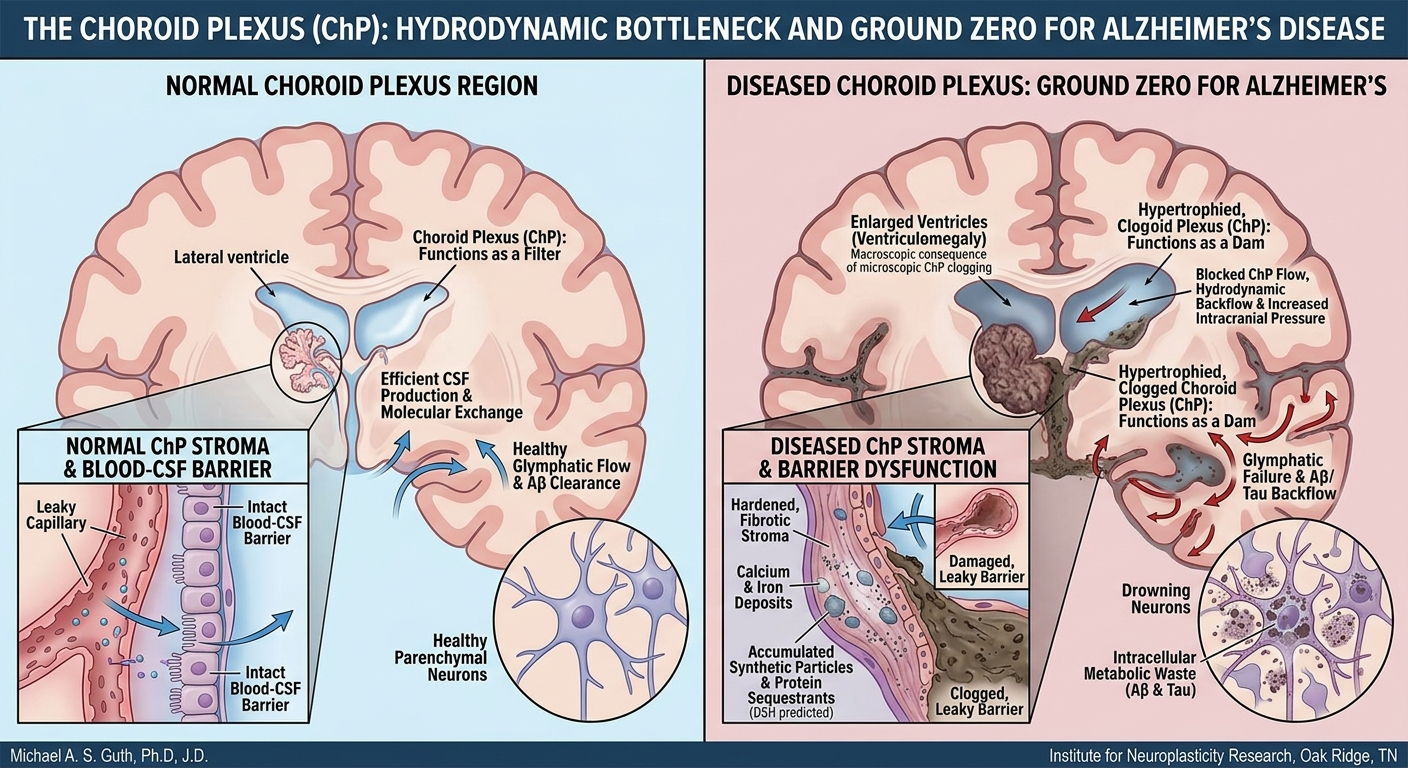
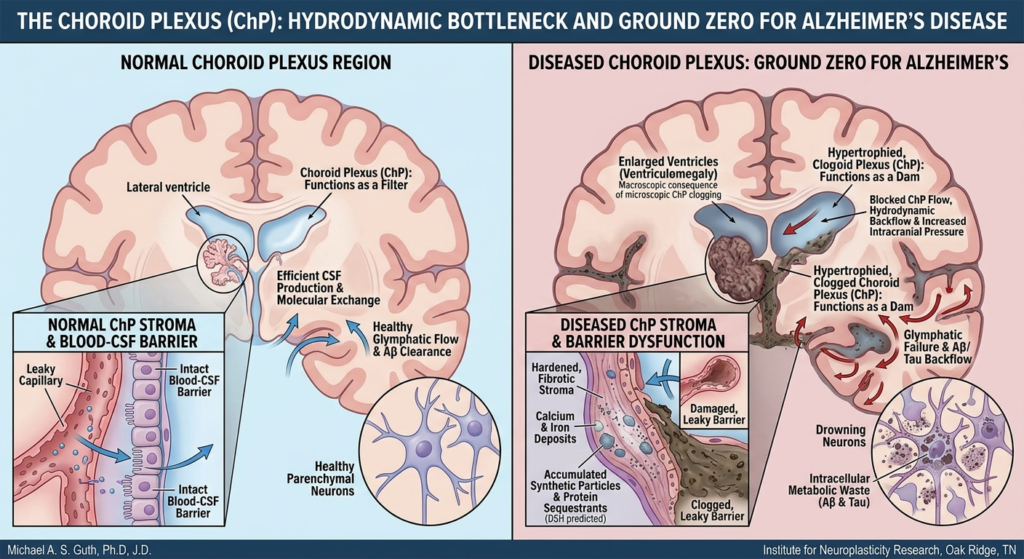
In this model, Aβ could be seen as an extracellular “sarcophagus,” a first-responder mechanism that encloses pathogens or insoluble or toxic material in the interstitial space, a role supported by its antimicrobial and metal-chelating properties (Atwood et al., 1998; Soscia et al., 2010). Tau, in turn, could function as an intracellular “lockbox,” attempting to isolate harmful material that has been internalized. These may represent protective, containment strategies.
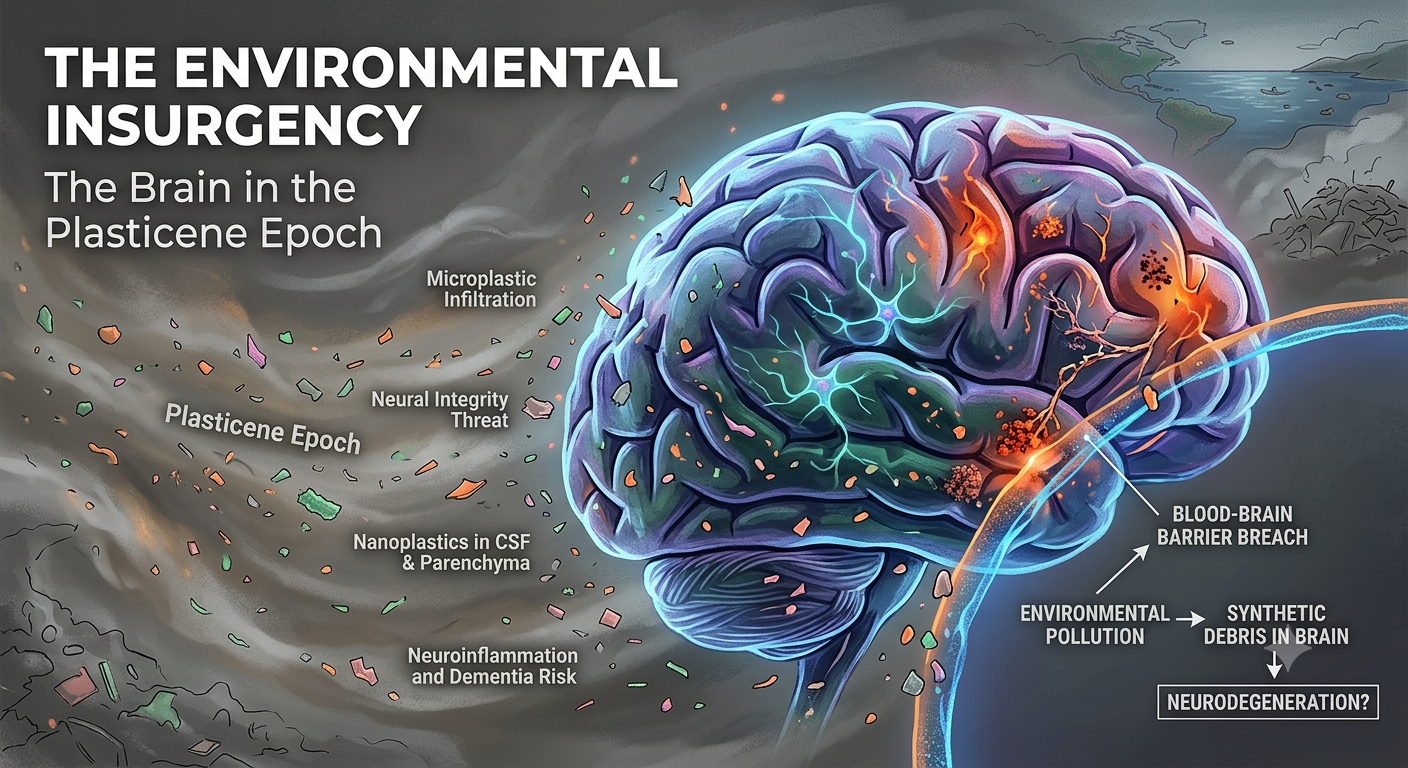
The catastrophic shift of the Plasticene era is the introduction of the indestructible synthetic polymer—NPs—which act as permanent, non-biodegradable nucleation seeds. These seeds hijack the ancient sequestration machinery, leading to the formation of permanent, enzymatically indigestible “synthetic-protein complexes” (Gou et al., 2024).
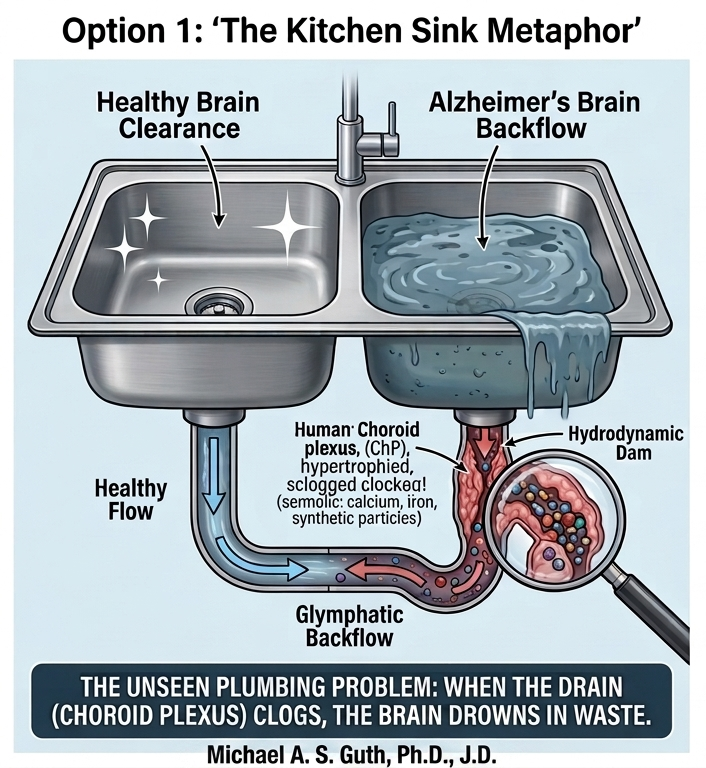
The DSH contends that disease progression occurs via a maladaptive phase transition from stable containment to lytic failure (Ferrer, 2022). The chronic burden of these indigestible complexes leads to microglial “immune frustration,” a metabolic and inflammatory tipping point. This state could be ignited by glutamate-mediated excitotoxicity, triggering microglial NLRP3 inflammasome activation and pyroptosis—a fiery, lytic cell death (Lassmann, 2022; Wang & Shen, 2024). Pyroptosis liberates the synthetic seeds, allowing them to propagate via the brain’s glymphatic drainage system, mechanically obstructing flow and seeding pathology in a pattern that recapitulates Braak stages (Iliff et al., 2012; Rasmussen et al., 2022).
This framework offers a direct explanation for the therapeutic paradox: mAbs remove the proteinaceous sarcophagus but leave the synthetic splinter exposed, causing inflammatory rebound (ARIA) and continued seeding. It shifts the etiological focus from the host’s response to the environmental trigger and the failure of the clearance systems meant to handle it. By integrating planetary-scale environmental change with molecular neuropathology, the DSH moves the field beyond the amyloid-tau cul-de-sac, offering a new mechanistic narrative for diagnosis, therapeutic strategy, and prevention.